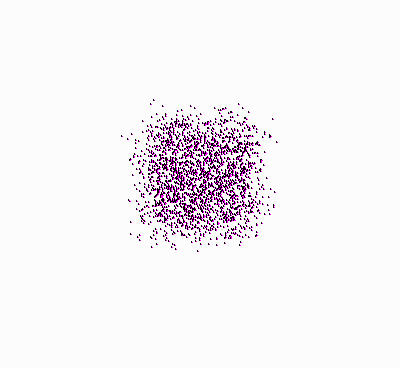Origin of the Second Law of Thermodynamics.
|
|
|
|
||||||||
| A hot piece of iron (red) is placed in a bucket with
cold water (black); after a while, the two come to a thermodynamic equilibrium. |
Molecules, starting at an orderly
structure (a crystal), and dispersing throughout the available space. |
The Contention
Scientists disagree on whether the two versions of entropy are related. Many of the well-known authors of books of physics that target a wide educated audience tacitly assume that the two notions of entropy are indeed related — actually coincide — so that speaking about the one is like speaking about the other. Such authors don’t even bother to make any distinction between the two: they talk about “order” and “disorder” (configurational entropy) in the context of the second law of thermodynamics (where talk about thermodynamic entropy should be appropriate, if they perceived a difference between the two). For example, according to Stephen Hawking:
“The nondecreasing behavior of a black hole’s area was very reminiscent of the behavior of a physical quantity called entropy, which measures the degree of disorder of a system. It is a matter of common experience that disorder will tend to increase if things are left to themselves. (One has only to stop making repairs around the house to see that!) One can create order out of disorder (for example, one can paint the house), but that requires expenditure of effort or energy and so decreases the amount of ordered energy available.
A precise statement of this idea is known as the second law of thermodynamics. It states that the entropy of an isolated system always increases, and that when two systems are joined together, the entropy of the combined system is greater than the sum of the entropies of the individual systems. For example, consider a system of gas molecules in a box. The molecules can be thought of as little billiard balls continually colliding with each other and bouncing off the walls of the box. The higher the temperature of the gas, the faster the molecules move, and so the more frequently and harder they collide with the walls of the box and the greater the outward pressure they exert on the walls. Suppose that initially the molecules are all confined to the left-hand side of the box by a partition. If the partition is then removed, the molecules will tend to spread out and occupy both halves of the box. At some time later they could, by chance, all be in the right half or back in the left half, but it is overwhelmingly more probable that there will be roughly equal numbers in the two halves. Such a state is less ordered, or more disordered, than the original state in which all the molecules were in one half. One therefore says that the entropy of the gas has gone up.” [In A Brief History of Time,[1] p. 106.]
Similarly, according to Brian Greene (emphasis in the original):
“First, entropy is a measure of the amount of disorder in a physical system. [...] Second, in physical systems with many constituents [...] there is a natural evolution toward greater disorder, since disorder can be achieved in so many more ways than order. In the language of entropy, this is the statement that physical systems tend to evolve toward states of higher entropy.” [In The Fabric of the Cosmos,[2] p. 154.]
and:
“The tendency of physical systems to evolve toward states of higher entropy is known as the second law of thermodynamics.” [ibid,[2] p. 156.]
Or, consider Paul Davies, another well-known and popular author:
“The so-called second law of thermodynamics is often phrased by saying that every closed system tends towards a state of total disorder or chaos. [...] One measure of the remorseless rise of chaos uses a quantity called ‘entropy,’ which is defined to be, roughly speaking, the degree of disorder in a system. The second law then states that in a closed system the total entropy can never decrease; at best it remains the same. Almost all natural changes tend to increase the entropy, and we see the second law at work all around us in nature. One of the most conspicuous examples is in the way that the sun slowly burns up its nuclear fuel, spewing heat and light irretrievably into the depths of space, and raising the entropy of the cosmos with each liberated photon. Eventually the sun will run out of fuel and cease to shine. The same slow degeneration afflicts all the stars in the universe. In the mid-nineteenth century, this dismal fate came to be known as the ‘cosmic heat death.’” [In About Time,[3] p. 34.]
As we see, well-known authors and scientists do not mince their words when it comes to identifying the increase of entropy with the increase of disorder. Notice how Davies, in the last excerpt, passes in the same breath from the configurational version that sees entropy as disorder to the thermodynamic version that sees entropy as “heat death” (essentially as the loss of ability to produce useful work).
However, some other, not-equally-well-known authors prefer to see an unbridgeable gap between thermodynamic and configurational entropy. They say the second law is about thermodynamics, and “thermo” means “heat”, don’t forget that folks! They think the observation that both configurational and thermodynamic entropy tend to increase is a curious coincidence, a mere analogy, and that we shouldn’t read too much into this analogy. They point out that the thermodynamic entropy is measured in specific units; namely, joules per degrees Kelvin, whereas the configurational one is a mere number, with no units attached. (The configurational entropy is the logarithm of all possible arrangements — e.g., of molecules — that result in indistinguishable configurations; therefore, it is a pure number.) Then they also note that increase in thermodynamic entropy reduces our ability to do useful work (e.g., to power a machine), but there is no such notion in the case of configurational entropy.
Most vociferous among those scientists has been Frank L. Lambert, a retired Professor Emeritus of Chemistry at Occidental College, Los Angeles, California, who has literally filled every nook and every cranny that concerns the 2nd law of thermodynamics on the web, “fighting” against the idea that the thermodynamic and configurational entropies are related. According to this Wikipedia article, “He is known for his advocacy of changing the definition of thermodynamic entropy as ‘disorder’ from US general chemistry texts and its replacement by viewing entropy as a measure of energy dispersal.” The following excerpt from the Wikipedia article on the second law of thermodynamics looks as if it’s taken directly from Professor Lambert’s own web pages:
“[T]he concept of entropy in thermodynamics is not identical to the common notion of ‘disorder’. For example, a thermodynamically closed system of certain solutions will eventually transform from a cloudy liquid to a clear solution containing large ‘orderly’ crystals. Most people would characterize the former state as having ‘more disorder’ than the latter state. However, in a purely thermodynamic sense, the entropy has increased in this system, not decreased. The units of measure of entropy in thermodynamics are ‘units of energy per unit of temperature’. Whether a human perceives one state of a system as ‘more orderly’ than another has no bearing on the calculation of this quantity. The common notion that entropy in thermodynamics is equivalent to a popular conception of ‘disorder’ has caused many non-physicists to completely misinterpret what the second law of thermodynamics is really about.”
Well, if professor Lambert is right, in addition to the non-physicists, it looks like a few true physicists (including such luminaries as Stephen Hawking, Brian Greene, and Paul Davies) also managed “to completely misinterpret what the second law of thermodynamics is really about”, whereas Prof. Lambert (a chemist) understood more deeply entropy and the second law. Could that be true?
But Professor Lambert is far from being alone in his rejection of the connection between thermodynamic and configurational entropy. There are numerous other scientists agreeing with him, as his successful campaign to “cleanse” American chemistry textbooks from the “blemish” of this “confusion” shows. Yet others draw quite different conclusions starting from the assumption that thermodynamic and configurational entropy are unrelated. For example, Brig Klyce, in this web page, argues as follows: he agrees that the Earth is not a closed system thermodynamically, since it receives an influx of energy from the Sun, but he says it must be a configurationally closed system (i.e., as far as order is concerned), since thermodynamic and configurational entropy (or: energy and order) are not related: the influx of energy cannot cause an increase in order. So Klyce tries to explain the increase in biological order on Earth, and claims that it must have some origin other than the energy received from the Sun. He uses this argument to support the notion of “panspermia”, the hypothesis that life did not evolve on Earth, but was “injected” here from the outer space (a theory that leaves unanswered the question of where and how life appeared first, but that’s another issue). He concludes that life has received such “injections” of order from outside the Earth, and that’s why it appears highly ordered. His view, of course, can also be used to support the creationist view that life did not evolve by means of natural selection, but was created on earth by some “Intelligent Designer”. (Klyce admitted to me that much, in personal communication.) So we see that the contention over the relatedness between thermodynamic and configurational entropy has ramifications beyond physics.
The Resolution
In what follows I will show that the two versions of entropy are consequences of a deeper mathematical — actually statistical — result, where “deeper” does not necessarily mean “harder to understand”; it’s actually quite simple to grasp. So the Hawking-et-al. view will be vindicated, whereas the drive to rewrite American chemistry textbooks will prove meaningless, a mere exercise in futility. But before stating a mathematical theorem and proving it, I prefer to motivate the reader appropriately with an observation.
Let’s take another look at the figure showing
the molecules that disperse in space (once again, press
![]() ):
):
|
|
What do we have here? Are these real molecules that disperse in space?
Of course not. These are just some simulated
“molecules”, mere circles painted in various colors, moved
around by a program that runs when you load this web-page and
press the button
![]() .
.
In fact — wait a minute: these circles are not even moved around! Come to think of it, nothing moves around here. The screen of your computer contains some pixels, and the program sometimes paints some of them in various colors, other times it paints the same pixels black, and so on. Pixels don’t move around on your screen, they just are, at fixed locations. The program tricks you into believing that circles move around by painting pixels in different colors, at appropriate times.
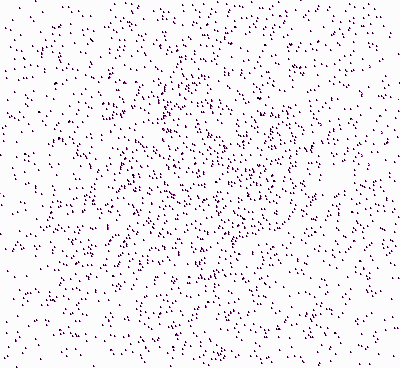
But I swear, I didn’t do anything in the program with the explicit purpose to trick you into believing that there are circles that disperse within the black rectangle! All I did was that I asked the program to draw a colored circle around each point, first arranging points in an orderly fashion (100 points in a 10 x 10 matrix), and then moving each point individually, independently of where the other points are, to a neighboring location. (“Neighboring” means a location at a fixed distance from the given one, at a random angle; thus the new location is anywhere on the circumference of a circle centered at the previous location and with radius a given fixed distance.) I didn’t give any explicit command to points to “disperse”; there was no order to them to “get away from the center of the area”. And yet they disperse, just like real gas molecules in a real room! But why? What is the connection? Okay, the molecules are physical things, they carry their mass around in actual space, and we could try to examine why they disperse by applying the known laws of physics. But these points? Why on earth do they disperse as well? We have no physics to work here with, it all happens in virtual “space”, in a computer. Of course, one can claim there is something physical that implements those points and paints them as colored circles: it’s the bits in the chips of your computer’s memory. Sure, but the relation between points/circles and bits in computer chips is so tenuous that you can’t figure out which bits do what. As a matter of fact, bits do not disperse in your computer’s chips. Moreover, your computer and its hardware are entirely irrelevant, because this program could be run on a Turing Machine, abstractly, mathematically, as any first-year graduate student of computer science will assure you. Something else is going on here, which has nothing to do with physics: there are neither real molecules in space, nor red-hot pieces of iron cooling down here. We have a third example of dispersal above, a third kind of entropy, independent of the other two physical ones.
Conclusion:
| Something like entropy and the “second law of thermodynamics” can be observed even abstractly, mathematically, without any material implementation, with no physical substructure to make it real. |
Hmm... Too many “entropies”, don’t you think? Too many to be all just analogies of each other, by mere coincidence. We should become suspicious with so many “coincidences”, and look for something deeper that underlies and unifies them all. And because this third kind of “entropy” is not even physical (should we call it “virtual entropy”?) we might as well forget for a moment about physics, and seek to explain this third kind first, because it seems “purer”, without any of the material baggage of the other two cases. After we explain how this “virtual dispersal” happens, we can see how it acquires physical implementations that explain the thermodynamic and configurational cases of entropy.
So, let’s see: what we want to explain is why a given point in space (one of those circles) that performs a “random walk” moves away from a given original location on average. This last qualification is very important: it must be an average distancing from the original location, because a random walk implies that the point might come back to its original location, for all we know. But if we repeat the experiment a large number of times, and average out the locations of the point at each time-step, we should find that the point increases its distance from its original location. But why? Well, here is a simple qualitative explanation:

The above figure shows a point marked with a purple dot at the coordinates (x, y). The point is supposed to have started its random walk at the origin, marked with (0, 0), and is now “contemplating” to jump randomly anywhere on the circumference of the black circle, because that’s how it moves: at any time it is at some location (x, y), and at the next time-unit it finds itself somewhere at a distance of 1 space-unit away from (x, y), having moved at a random angle. Here is the crucial question: Although all destinations on the circumference of the black circle have an equal probability to receive the point (that’s a given), how many of them imply that the point will move away from (0, 0), and how many that it will move closer to it? The following figure answers this question:
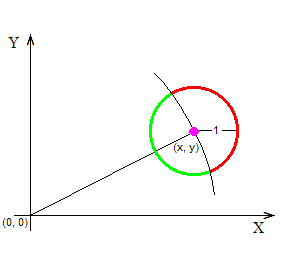
Those destinations that imply that the point (x, y) will move farther away from the origin (0, 0) have been marked in red color; and those that imply that (x, y) will move closer to (0, 0) have been marked in green color. It’s not that the red points are “more” than the green ones (they are both infinite in number), but the length of the red arc is longer than the length of the green arc, and so (x, y) has greater probability to end up on the red (away) rather than on the green (closer) part of the colored circle.
The above is the qualitative explanation, but there is also a quantitative one. We can compute how fast the point (x, y) will distance itself from the origin (0, 0) on average, assuming the above “rules of motion”, provided we make things formal. However, if the reader feels uncomfortable with formulas and math, please note that there is nothing essential to be missed if the proof of the following theorem is skipped; just make sure you read the theorem itself and understand its statement, because it is of central importance in this whole discussion.
Definition. A 2D random walk is an infinite sequence of points p0, p1,... on the 2-dimensional Euclidean plane such that each point pk has a distance of 1 from its previous point pk–1, for all k > 0. Point p0 is called the origin of the walk, and point pk is called its k-th step.
The definition implies that we don’t care about the direction of the straight line defined by points pk and pk–1, therefore the direction of this line is random; hence the term “random walk”.
Theorem (“of dispersion
in 2D space”). The expected distance between the origin p0 and the n-th
step pn of a 2D random walk is equal to ![]() . (Here, “expected distance” refers to
the mean value of distances from the origin p0 of the n-th
steps of a large number of 2D random walks that share a common
origin p0.)
. (Here, “expected distance” refers to
the mean value of distances from the origin p0 of the n-th
steps of a large number of 2D random walks that share a common
origin p0.)
Proof: Think of the 2D plane as the plane of complex numbers, and place the origin p0 at (0, 0). Now let (x, y) be the coordinates of the n-th step pn in a 2D random walk. Therefore, as a complex number z, point pn would be written as z = x + iy. Recall that each complex number x + iy can also be written using Euler’s exponential notation as follows:
z = x + iy = |z| (cos(u) + i sin(u)) = |z| eiu
where |z| is the modulus, or distance of z from the origin p0 = 0 + i0, and u is the phase, or angle between the x-axis and the line that connects z with the origin p0.
Now, if we fix the modulus |z| to the value 1 (since point z = pn differs by this fixed distance from its previous point pn-1 in the random walk) and allow the phase u to vary randomly in the interval [0, 2π) (since the angle between successive points is arbitrary), then the new position z of (x, y) after n steps on the complex plane must be given by the following sum:
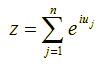
Recall that the absolute square |w|2 of a complex number w (i.e., the square of its distance from the origin) is equal to w·w΄ where w΄ is the conjugate of w, i.e., w΄ = x – iy = |w| e-iu. So the absolute square of z in the above formula, which is equal to z·z΄, is given by:

Now, let’s try to compute the mean value of the quantity |z|2. We shall use angle brackets (< >) to denote mean values. We have:
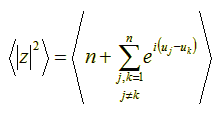
Since both angles uj and uk are random variables with identical means, their difference (uj – uk) is also a random variable with mean 0 (zero). This means that the whole formula of the mean value to the right of the plus sign, above, is 0 (zero). Thus, simplifying we get:
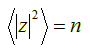
Hence, taking the square root on both sides, we find that the root-mean-square distance |z|rms after n unit steps is:
![]()
The root-mean-square is the
average distance of z (or point pn) from the
origin p0, which is what we wanted to show. ![]()
If n
represents time in the above calculations, then the
theorem tells us that at time n the point that performs
a random walk is expected to be found at an average distance of ![]() from the origin (point
of departure). This result is actually the same as found by
Einstein in his famous papers of 1905 and 1906 on Brownian
motion,[4] except that Einstein’s calculations and notation are
much harder to follow. The notation used in the above proof was
taken from Weisstein, 1997 (p. 1524).[5]
from the origin (point
of departure). This result is actually the same as found by
Einstein in his famous papers of 1905 and 1906 on Brownian
motion,[4] except that Einstein’s calculations and notation are
much harder to follow. The notation used in the above proof was
taken from Weisstein, 1997 (p. 1524).[5]
The above “dispersion theorem” tells us why the circles that perform random walks and are drawn by the program disperse on average, even though the program does not give them any explicit command to do so. The theorem explains what I earlier called “virtual entropy”.
Now it’s not difficult at all to see how the configurational and thermodynamic dispersals (and their associated notions of entropy) are simple physical implementations of the above mathematical result.
The configurational case is
trivial to see. We assumed molecules of a gas in a container
covered with a lid, placed at the center of an isolated room that
contains a different gas. Suppose the pressure in the two gases
is identical. The molecules of the gas in the container (as well
as those outside) perform approximations of random walks as they
meet and bounce off one another. When we open the lid of the
container the molecules of its gas keep performing random walks,
but now they are permitted to meet and bounce off any kind of
molecule: either of the same gas, or of the gas outside. So they
disperse in space just as the colored circles in our simulation
do, for the statistical reason explained in the dispersion
theorem, even though they don’t move at a fixed distance every
time, and even though they are not restricted to move on a 2-D
plane. (What was proven by the theorem in two dimensions holds
also in three dimensions, except that the average speed of
dispersal in not equal to ![]() but slower. The proof is considerably more complex,
that’s why it was given in two dimensions and with a fixed
distance of movement.)
but slower. The proof is considerably more complex,
that’s why it was given in two dimensions and with a fixed
distance of movement.)
People often become confused with
variations in the physical details of this experiment. They
may consider the room empty of matter (a vacuum), so when the lid is
opened the molecules of the gas in the container swoosh out and
quickly spread throughout the room without performing random
walks. Okay. The molecules of a gas under normal temperature move
at dizzying speeds all the time anyway. (According to
this article by Prof. Lambert, “At the usual lab temperature, most
water molecules are moving around 1000 miles an hour, with some
at 0 and a few at 4000 mph at any given instant.”) We are not
aware of their speeds under normal pressure because they meet
other molecules and bounce off before they have a chance to go
too far. If, however, they find that their way is free of
obstacles (as in a near-vacuum), then of course they’ll rush
unhindered at their dizzying speeds, and that’s what will cause
the swooshing in the void of the room when the lid is opened.
There is nothing mysterious to explain here. Practically the same
phenomenon appears when you open a soda can: the great difference
in pressure between the molecules of CO2 in the can (highly pressurized) and the molecules of
air outside (in lower pressure) causes the CO2 molecules to swoosh
out of the can and disperse in the air (especially if you have
shaken the can and thus increased the internal pressure, hence
the speeds of the CO2 molecules). Same phenomenon. The vacuum, or the low
pressure, simply speeds up the rate at which the molecules
disperse. The following simulation shows precisely that effect (click on
![]() ):
):
|
|
Here, the simulated molecules make longer jumps before they bounce, as they would if they could move in a relatively empty space. The result is the “explosion” that you see when you run the program, and the much faster filling up of the available space.
Now let’s turn our attention to the thermodynamic case (red-hot piece of iron thrown in cold water), because that is what professor Lambert and others dispute as having any relation with the case of configurational entropy.
When a hot piece of iron is placed into cold water the molecules of the water “acquire energy” (we’ll see what that means) and vibrate more vigorously. But because they are molecules in a liquid, they can disperse in the volume of the rest of the water. I prefer to avoid this dispersal of molecules in our thought experiment, because I want the thermodynamic case to appear as different as possible from the previous experiment with the gas molecules in a container that disperse in a room. So I’ll propose a small modification in our thermodynamic setup — an improvement, actually. Suppose that instead of water we have a piece of concrete. This concrete chunk has a square-shaped hole at its center, and instead of the “hot iron” we have another piece of concrete, but hot, which goes and fits neatly and perfectly(*) into the square hole. Hot concrete goes and fits into the hole of cold concrete, that’s all I’m saying. (Our figure, below, remains identical: suppose the black area is the cold concrete, and the red is the hot piece.) Now there can be no dispersal of molecules, and yet the second law of thermodynamics guarantees that the smaller hot piece will cool down, the larger surrounding chunk will warm up, and the two will come to a point of thermodynamic equilibrium after a sufficiently long interval of time. The earlier figure is repeated below, for our convenience.
|
|
Let’s think now: how does that happen? What is the mysterious “energy” that flows out of the hot piece? What does it consist of? How does it flow and why does it disperse?
In this section we’ll see the correct explanation of how “energy” (or “heat”) disperses, and in the next section we’ll review some bogus explanations that have been proposed by others.
When the red-hot piece of concrete is placed in the square hole in the middle of the cold chunk of concrete, what kind of interaction can occur between the two bodies at the molecular and quantum level?
Well, what we know is that some of the vigorously vibrating molecules of the red-hot piece (those that are at its outermost region, its periphery), come into “contact” with molecules of the cold piece. But “contact” is not the right term when we talk about molecules, since “touching” is something that makes sense only at the macroscopic level. At the microscopic level, the atoms of some of the “hot” (vigorously vibrating) molecules will approach the atoms of some of the “cold” (less vigorously vibrating) molecules.
Fine, so atoms will approach atoms. Again, atoms cannot “touch” each other microscopically, so when we speak of an “approaching” at the atomic level we mean that the electrons of the outermost shells of those atoms will come close together (always while in vibration). So, what happens when electrons approach electrons?
Quantum theory says that electrons that come close together exchange virtual photons. If the electrons were free in space, they would scatter at random angles after this exchange. Now that they are bound to the nuclei of atoms by means of the electromagnetic force, most probably they will continue being bound to their atoms, but their mutual bouncing off will cause their respective atoms to bounce off, too. (This mutual repelling due to the electromagnetic force is the reason that solid objects like ourselves stay on top of other solid objects, like chairs and floors, and do not pass through them.) What interests us is that because one of the two electrons (assume only two of them interacting, for simplicity) moves faster in space than the other one (because it follows the vibrations of the atom it belongs to, which is “hotter” than the other one), quantum theory says that there is a higher probability that virtual photons will go from the fast-moving electron to the slow-moving one, so that the former will lose some of its energy, whereas the latter will gain some. As always in the quantum world, we talk about probabilities, not deterministic events. But what concerns us is the average case, and on average the fast-moving electron will send one or more virtual photons to the slow-moving one. This is one mechanism by which the mysterious “energy” is transmitted from one material to the other: this kind of energy is a flow of virtual photons, which cause the electrons — hence their respective atoms — to recoil. But there is more.
The hot piece is depicted in red color on our drawing, right? And in reality, red-hot things are, well... red. They are red for the following reason. Their highly excited electrons spontaneously drop to lower-energy levels, emitting photons as they do so. (Real photons, not virtual ones.) The more excited the electron, the higher the probability that the photon will have a small wavelength (high energy). Higher energies mean photons with wavelengths possibly in the visible range, such as in the red part of the electromagnetic spectrum (and even further toward the orange and smaller wavelengths, depending on how hot the object is). As the hot body cools down (we haven’t seen yet how), the emitted photons are of longer wavelengths, and so are shifted toward the infrared. That’s why the red-hot piece becomes first dull-red as it cools down, then its color fades more, until essentially all the emitted photons are in the infrared, so we don’t see them anymore (consequently we see the “natural” color of the body at lower temperatures, be it black or gray, whatever is reflected by ambient light).
As I mentioned, these emitted photons are not virtual but familiar photons that would be registered by our retinas if we could see them. But we can’t see them because they are in the closed system of the two concrete pieces of our experiment. So they are emitted within the molecules of the material, and can’t go too far because these are chunks of concrete we are dealing with, and concrete is opaque to light (less so to infrared photons, but still relatively opaque). So the emitted photons travel short distances before being absorbed by electrons of neighboring molecules, which they might excite and cause to jump to higher energy levels. Then those excited electrons might emit a photon again and drop back to a lower energy level. The photons are emitted at random angles. So, although we can’t talk about “the same” photon moving from molecule to molecule (or from atom to atom, or from electron to electron), the net result of all this is that there is something like a random walk of photons.
Hmm... a random walk. This is clearly the case with normal photons. But the virtual photons, too, do something analogous, because they, too, are exchanged between electrons at random angles.
So this is what the mysterious “flow of energy” is: it’s photons that perform random walks. “Energy” in the context of our experiment is not a substance made of some mysterious and otherworldly material, but a convention for the wavelength of photons: the shorter the wavelength, the higher the energy. At a macroscopic level it’s often useful to model energy by the quantity of an abstract “substance” (e.g., “temperature”), because this allows us to solve conveniently problems about objects and processes in the macro-world. But down at the microscopic level of description, in our thermodynamic experiment, energy is associated with the wavelength (or frequency) of photons, i.e., photons that, as I said, perform random walks (or behave as if the “same” photon performs a random walk).(*)
Therefore, random walks are the reason why energy disperses within the cold body until there is an equilibrium, and the dispersion theorem models the situation. And this discussion tells us that the two figures that were juxtaposed at the top of this page essentially simulate the same phenomenon: the figure on the left shows the situation macroscopically, with photons performing random walks and dispersing within the material; and the figure on the right shows the situation microscopically, depicting individual molecules of a fluid that perform random walks and disperse in a box. In the latter case, what we have is the dispersal of matter. In the former case we are tempted to say that we have the dispersal of “energy”; but the reason I keep putting the word “energy” in quotes is because I want to emphasize that even in this case we still have the dispersal of matter. For, is a photon immaterial? Of course not, it’s a little lump of “matter” in the generalized sense, the sort of massless matter that we prefer to identify with energy (but which can be assigned a non-rest mass m through the relation m=E/c2, where E is the energy of the photon). The useful distinction that can be made is that the configurational case involves the dispersal of matter in the form of mass, whereas the thermodynamic case involves the dispersal of matter in the form of energy. But, by whatever name and form it goes, generalized “matter” disperses in spacetime when its quanta perform random walks, and the reason is not physical, but mathematical, given by the dispersion theorem. Note please: when I say “the reason is not physical” I don’t mean it’s supernatural! I mean that the laws of physics as we know them (including Newton’s laws of motion, quantum mechanics, and everything we know about the four forces of nature), do not suffice to explain the reason for the average dispersal of random-walking particles. An extra-physical, a mathematical result is needed to explain this phenomenon. Of course, if we include this mathematical result into the notion “laws of physics”, then we can again say that the laws of physics — in this inclusive sense — fully explain the phenomenon.(*)
I hope the above discussion explains sufficiently the claim that I made earlier: the two entropies, thermodynamic and configurational (and even the third kind that I briefly referred to as “virtual”, earlier), are manifestations, or implementations, of a deeper mathematical result. The “second law of thermodynamics”(*) is, similarly, at work whether we talk about energy or mass dispersing in spacetime. Consequently, the drive to eliminate references to configurational entropy in American chemistry textbooks is utterly meaningless and even short-sighted, because it disseminates knowledge superficially to students, who can’t see the deeper mechanism that’s responsible for material dispersal, but see only one aspect of it: the one that appears in the thermodynamic case.
However, although the relation between thermodynamic and configurational entropy is unassailable, there is still confusion about the role of order and disorder in this context.
The Confusion
Let us see a couple of explanations for the second law of thermodynamics that have been given, which ignore the dispersion theorem and the deeper relation between thermodynamic and configurational entropy. First, consider the configurational case, and Brian Greene’s explanation for why molecules (or other material things in general) disperse in space.
Greene first presents the notion of entropy by asking the reader to imagine tossing an unbound copy of Tolstoy’s War and Peace high into the air, letting the 693 double-sided loose pages drop on the ground, and then collecting them one by one, without looking at their page numbers. What is the probability that the pages will be collected in their correct order? Greene calculates that there are about 101878 different out-of-order page arrangements (and presents the entire 1878-digit number using the better part of p. 152 of his book). He observes that there is only one “correct” (or desired) order, so the probability to pick up the pages in the correct order and keep reading about Anna Pavlovna and Nikolai Ilych Rostov (and understanding what is being read) is about 1/101878, i.e., vanishingly small. So far so good.
Now let’s see how he explains a more typical experiment that is often described in the context of entropy and the second law of thermodynamics. Consider opening a bottle of Coke. Gas, like CO2, is initially confined in a small space in the bottle. After we open the cap of the bottle, the molecules of CO2 spread evenly in the room. Here is how Greene explains this:
“[W]hen you twist off the bottle’s cap [...] you open up a whole new universe to the gas molecules, and through their bumping and jostling they quickly disperse to explore it. Why? It’s the same statistical reasoning as with the pages of War and Peace. No doubt, some of the jostling will move a few gas molecules purely within the initial blob of gas or nudge a few that have left the blob back toward the initial dense gas cloud. But since the volume of the room exceeds that of the initial cloud of gas, there are many more rearrangements available to the molecules if they disperse out of the cloud than there are if they remain within it. On average, then, the gas molecules will diffuse from the initial cloud and slowly approach the state of being spread uniformly throughout the room. Thus, the lower-entropy initial configuration, with the gas all bunched in a small region, naturally evolves toward the higher-entropy configuration, with the gas uniformly spread in the larger space.” [In The Fabric of the Cosmos, pp. 155–156.]
But there is a glitch in the above explanation. Okay, the initial gas cloud is confined in a smaller space than the space of the entire room. But why would a molecule choose to move away from the cloud? What pushes it there, to the rest of the room? One might counter, the highly pressurized state of the gas in the bottle pushes it, i.e., the vigorous bumping and jostling with the other molecules of the gas cloud. Yes, but we don’t have to imagine a pressurized gas. The same phenomenon will be observed if the pressures of the gases inside and outside the bottle are identical: still the gas-inside will spread in the rest of the room. So, again: what “pushes” molecules to “explore” the rest of space, as Greene puts it, even under a lack of pressure differential? Merely because “there are many more rearrangements available to the molecules if they disperse out of the cloud than if they remain within it” is not an explanation, because molecules don’t care about numbers of rearrangements and “opportunities” given to them to “explore” some terra incognita; they merely move randomly in space! For all we know (if we ignore the dispersion theorem), they could be roaming forever around their original location, and thus staying within the gas cloud. Greene comes close to the qualitative explanation given earlier on this page (I’m referring to the figure of the colored red-and-green circle, just before the theorem), but he doesn’t quite put his finger on it.
Greene gets distracted (and distracts the reader) with the pressure differential, instead of concentrating on an example with a lack of such differential. If I shoot a bullet with a gun against a target, it should be of little wonder if I see the bullet hitting the target; but if I simply take a bullet in my hand, then let go, and see it floating away from my fingers, then I am confronted with a phenomenon that requires a nontrivial explanation.
Next, consider the thermodynamic case and Prof. Lambert’s crusade to “purify” American chemistry textbooks from the configurational “blemish” by severing the relation between thermodynamic and configurational entropy, making reference only to the thermodynamic case in serious, scientifically approved textbooks. Lambert attempts to explain the second law of thermodynamics (i.e., the statistical increase in thermodynamic entropy) in this web-page by means of a dialog between a Professor (presumably himself) and an imaginary Student (perhaps his alter ego: a bombastic individual that should serve as an example of how students should not behave if they’re really interested in learning, as opposed to showing off their knowledge). Here is the Professor’s explanation of the microscopic reasons why energy disperses in space.
First, the Professor observes that molecules of water (he uses water as a typical example) move in three different ways; or, better stated, their motion has three degrees of freedom, or components: a translational component by which they change their location in space; a rotational component, by which the entire H2O molecule rotates around some axis (which can change in time); and a vibrational component that concerns the bonds between O–H atoms within the H2O molecule, by which the distance between such bonds periodically increases and decreases (extremely fast by our human temporal standards, of course). The following figure is from the above-referenced web page, and shows the three motional components for a water molecule.
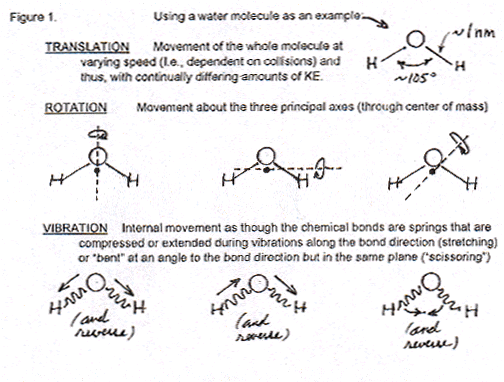
What the Professor never states explicitly in his discussion with the Student is the obvious observation that, of the three components of molecular motion, only the translational one can be suspected (held responsible) for the dispersal of energetic molecules in space. Clearly, no matter how fast a molecule rotates, or how fast the bonds between its atoms vibrate, it will not be translated in space. Thus two-thirds of the Professor’s description of molecular motion are irrelevant for the purposes of explaining dispersal.
Then the Professor goes on to explain to the Student how each of the three motional components is quantized, i.e., there are only specific and discrete values for the angular momentum of the rotational component: it’s not that the molecule can change its rotational speed along a continuum of values. The same is true for the vibrational and translational components. The Professor observes that the available quantized (discrete) values of the translational component are way more (really-really way more) in number than the possible discrete values of the rotational and vibrational components. But since the latter two are irrelevant for the explanation of dispersal, I will ignore this distinction in the numbers of quantized values.
Now comes the crucial part in the Professor’s explanation.
He says, take a snapshot of the current state of motion of the water molecules. Call this a microstate. Thus, in a given snapshot, the translational component of molecule #477,275,846,375,832,218 has a certain value (I said I’ll ignore the other two components); its neighboring molecule #477,275,846,375,832,219 has a different translational component; and so on. Collect all those components for all the zillion molecules in the quantity of water under consideration, and you have your microstate. Here it is, in Professor’s own words:
Imagine that you could take an instantaneous snapshot of the energy of all the individual molecules in a flask containing a mole of gas or liquid at 298 K. Remember that each molecule’s energy is quantized on a particular energy level. Then, each of the far-more-than Avogadro’s number of accessible energy levels (at that temperature and in that volume) could have zero, one, or many many molecules in it or “on it”. The whole snapshot showing each molecule’s energy of that mole is called a microstate — the exact distribution on energy levels of the energies of all the molecules of the mole at one instant in time.
Now consider what will happen next (says the Professor). The first of the above molecules will bounce against another one (usually a neighboring molecule, since this is water and its molecules can’t move too far before being hit by others) and will change its translational component. But there are many available options for the new value of its translational component. And this is true for all molecules in the fluid. Therefore, over time, the molecules will “explore” the space (note: the abstract space, says I) of possible values of their translational components. Hence we’ll end up with microstates that have their translational space widely distributed, as opposed to the single initial microstate. In Professor’s words:
Since a collision between even two molecules will almost certainly change the speed and thus the energy of each one, they will then be on different energy levels than before colliding. Thus, even though the total energy of the whole mole doesn’t change — and even if no other movement occurred — that single collision will change the energy distribution of its system into a new microstate! Because there are trillions times trillions of collisions per second in liquids or gases (and vibrations in solids), a system is constantly changing from one microstate to another, one of the huge number of accessible microstates for any particular system.
Then the Student asks a crucial question:
What does “more microstates” for a system have to do with its energy being more spread out? A system can only be in ONE microstate at one time.
And the Professor answers as follows (my emphasis):
Yes in only one microstate at one instant. However, the fact that the system has more ‘choices’ or chances of being in more different microstates in the NEXT instant – if there are “more microstates for the system” – is the equivalent of being “more spread out” or “dispersed” instead of staying in a few and thus being localized. [...]
You [i.e., the Student] already stated the most important idea, a single microstate of a system has all the energies of all the molecules on specific energy levels at one instant. In the next instant, whether just one collision or many occur, the system is in a different microstate. Because there are a gigantic number of different accessible microstates for any system above 0 K, there are a very large number of choices for the system to be in that next instant. So it is obvious that the greater the number of possible microstates, the greater is the possibility that the system isn’t in this one or that one of all of those ‘gazillions’. It is in this sense that the energy of the system is more dispersed when the number of possible microstates is greater — there are more choices in any one of which the energy of the system might be at one instant = less possibility that the energy is localized or found in one or just a dozen or only a million microstates. It is NOT that the energy is ever dispersed “over” or “smeared over” many more microstates! That’s impossible.
So, what does “energy becomes more dispersed or spread out” mean so far as molecular energies are concerned? Simple! What’s the absolute opposite of being dispersed or spread out? Right — completely localized. In the case of molecular energy, it would be staying always in the same microstate. Thus, having the possibility of a huge number of additional microstates in any one of which all the system’s energy might be in — that’s really “more dispersed” at any instant! That’s what “an increase in entropy on a molecular scale” is.
So... let’s see. Suppose I have a marble ball in my hand, and there is a number of holes on the ground at a distance of about one yard from me. Each hole is large enough to let the marble fall inside, and suppose at most one marble can fit in each hole. I throw the marble forward, letting it roll on the ground toward the holes. Following the Professor’s logic (see the highlighted portion, above, and match it with what follows), although the marble can be in only one hole at one instant, because there is a large number of different accessible holes, there are a very large number of choices for the marble to be in that next instant. So it is obvious that the greater the number of possible holes, the greater is the possibility that the marble isn’t in this one or that one of all those holes. It is in this sense that the positional state of the marble is more dispersed when the number of holes is greater.
Does it make any sense? No, not to me. A marble can be in one hole at a time, period. How can its potential for “choosing” from among a large number of holes send it to more than one hole?
And if you think the analogy with a single marble (single molecule) is misleading, okay, think of 10 marbles. Throw them forward, as before. Aren’t they going to end up in exactly 10 different holes?
Now take those 10 marbles out of their holes, step back, and throw them forward again. Aren’t they going to end up again in exactly 10 different holes? 10 different holes, in general, to be sure. But why would the marbles disperse among the holes?
Unless, of course, we modify the marbles–holes experiment as follows: perhaps after we remove each marble from its hole we don’t step back, but place the marble just next to its hole, and give it a little kick toward a random direction. The marble then goes and falls into a neighboring hole. Then we continue like that, taking it out, and giving it another kick to a random direction. Then, the marble will perform a random walk, and the dispersal theorem tells us that if we repeat this many times with many marbles, on average the marbles will disperse.
But this modification cannot apply to the Professor’s energetic molecules, and here is why:
Each molecule has a given value for its translational component, right? That’s a given value for its kinetic energy level. (Note the “KE” in Prof. Lambert’s figure, above: it stands for “kinetic energy”.) So it is reasonable to imagine that any given molecule changes its energy level with each kick that it gets from other, neighboring molecules. Its energy value performs a random walk in the abstract space of quantized kinetic energy levels. But why would this kind of random walk in the abstract energy space imply that the molecule will perform a random walk in physical space? This is like saying that a person who is some times happier than other times (that is, performs a random walk in an abstract “happiness space”) is expected to visit more places in the world than another person who stays at the same happiness level all the time. Why? It doesn’t compute. It’s a non sequitur.
The translational components of the moving molecules can indeed acquire many different values, i.e., kinetic energy levels. But it is not “obvious” at all that this will cause the energy of the molecules to disperse in physical space — not unless we take into account the dispersal theorem, and the observation that, in the context of our experiment, energy is transmitted through photons, which are the agents that perform the random walks, from electron to electron. (It is interesting to note that the word “photon” does not appear even once in Professor Lambert’s “microworld explanation” of energy dispersal.)
What about order and disorder?
Prof. Lambert makes it very clear that all talk about order and disorder in the context of entropy and the second law of thermodynamics is wholly unjustified, an example of sloppy thinking, a “Cracked Crutch For Supporting Entropy Discussions”. This, in spite of the fact that well-known authors such as Hawking, Greene, and Davies (among others) seem to feel no compunction to talk in terms of order and disorder, as shown in the excerpts at the beginning of this page. Prof. Lambert begins as follows in the above-referenced article:
To aid students in visualizing an increase in entropy many elementary chemistry texts use artists’ before-and-after drawings of groups of “orderly” molecules that become “disorderly”. This has been an anachronism ever since the ideas of quantized energy levels were introduced in elementary chemistry. “Orderly–disorderly” seems to be an easy visual support but it can be so grievously misleading as to be characterized as a failure-prone crutch rather than a truly reliable, sturdy aid.
Prof. Lambert later proceeds with an example that, according to him, shows why the talk about order and disorder is a “cracked crutch”. He asks the reader to imagine a bowl with water and chunks of ice floating on its surface (figure below, on the left). After some time, the ice has melted and the bowl contains just water in liquid form (figure, on the right).

Ice floating on water in a bowl (left), and same bowl with plain water after ice has melted away (right)
People who are being introduced to the notion of entropy perceive the water-plus-ice-chunks as a disorderly collection of objects, says Prof. Lambert, whereas they perceive the later plain water as a uniform substance, an ordered form. So they might think we have a counter-example here: a disorderly collection of objects turned into an orderly soup. So they get confused (he claims).
Yes, I agree that people would get confused if what is order and what is disorder in a situation such as the above is described to them in the manner suggested by Prof. Lambert. But the Professor’s is a terribly misleading description. Learners are bound to be confused with misleading descriptions. A good educator must explain things in the right way.
The “order” in the bowl with the floating chunks of ice is to be found in the configurations of H2O molecules that form the ice crystals within each chunk of ice. There are many fewer configurations of molecules that form ice crystals in chunks (that’s “order”) than configurations of molecules that float free in the soup of pure water (that’s “disorder”, see more below). So, having heard the proper description, the learner will see an order-to-disorder progression and no contradiction with the 2nd law of thermodynamics. This is not a unique case of an initially wrongly formed perception due to intuition. Without proper tutoring in physics, people tend to think that heavy objects fall faster than light ones — even Aristotle was fooled on this one — that the Sun turns around the Earth once every day, and that mass is identical to weight. But a wrong first impression cannot be a reason for abandoning the more informed physical description.
Why do we say there is “more order” in the bowl with the floating ice, and “less order” (or more disorder) in the bowl with the plain water? Because the former situation is analogous to the molecules of a gas being restricted in a small volume, as in the examples discussed earlier in this text, whereas the latter situation is analogous to the gas molecules being spread out everywhere in the available space (see figure, below).
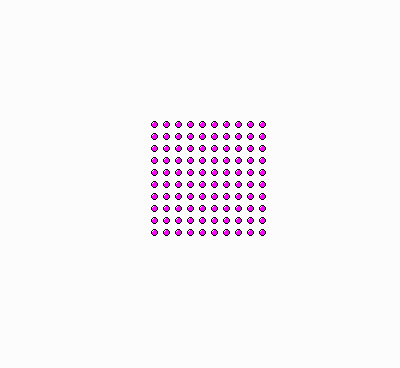 |
 |
|
| Ordered molecules, akin to ice crystals floating on water. | Molecules in disorder, akin to a “soup” of molecules in liquid water. |
There’s a subtle issue, however, when we say that the image on the left, above, is more “ordered”. The way I arranged the “molecules” (dots) in a square 10 x 10 grid, of course appeals to our sense of “order”. But why? How can we make the notions of “order” and “disorder” precise, so that even Prof. Lambert (and like-thinking scientists) will have no reason to claim that order/disorder is not an un-physical notion, a mere psychological thing, a “cracked crutch” for the understanding of entropy?
What can be made precise are not exactly the notions of order and disorder, but the very closely related notions of “compressible” and “incompressible” configuration.
The reason is this: to say that the molecules of the image on the left, above, are “ordered” we need the judgment of a person, who would notice that the molecules are arranged in a perfect matrix of 10 rows by 10 columns. I might have arranged the molecules in a diamond-like shape; or along the circumference of a circle; or make the 100 dots form 20 crosses of 5 dots each and place the crosses themselves on 4x5 matrix; or arrange them in an essentially unlimited number of other ways. In each such case, a person with enough intelligence and patience might see the pattern, and come up with a short description of the 100 dots. The person might, or might not see the pattern of dots that I selected, depending on how difficult the pattern is. In some difficult cases the person might fail to discover the pattern. Consider for example the picture on the right, above. It might be that I placed the dots on such x, y integer coordinates that the expression xy+1 is a prime number. I didn’t, but I could have done so. And nobody can guarantee that a person would succeed in discovering that relation.
If a relation for the position of dots (a pattern) is discovered, we say the configuration can be compressed. The notion of compression refers to the fact that the configuration of dots can be described in a short way (e.g., “20 crosses of 5 dots each”); otherwise we say the configuration is incompressible.
But the subtle issue is: if it is incompressible, is it so because nobody succeeded in compressing it (although there might be some yet unknown way), or because there really isn’t any way to compress it, even if “God” — so to speak, i.e., a super-intelligent being — attempted to do it?
The notion of incompressible information is directly related to the notion of randomness. We can think that the dots on the right-side figure, above, are randomly placed. Disordered, randomly placed, incompressible — all these words seem to refer to the same concept. But some mathematicians, physicists, and philosophers, would say that randomness (a.k.a. incompressibility, a.k.a. disorder), cannot be defined, because we can never know if a configuration of things (dots on the plane, numbers in a sequence, etc.) defies any description via a rule (making it patterned, or compressed, or non-random, etc.), or it just happens that no human being (or computer algorithm, etc.) has succeeded in compressing it yet. Since we cannot always know, they say, we don’t have a definition. For instance, the physicist Heinz Pagels says the following:
Andrei Kolmogorov, the great Soviet mathematician, thought he could define randomness by the criterion that if it took as long to state the rule, suitably transcribed into numbers, for the construction of the numerical sequence [as] the actual length of the sequence, then the sequence was “random.” However, finding the construction rule for the sequence depends on human cleverness, and we can never be assured that the rule we have found is the simplest one that gives the sequence. [...] A precise definition of randomness for finite sequences simply does not exist. [In The Cosmic Code,[6] pp 86–87.]
And yet, I want to make a proposal for avoiding this trap (the trap of not knowing whether a human might succeed compressing the configuration), and propose an objective mechanism that can serve as a definition of randomness (and of incompressibility, and of disorder, etc.).
Take a very specific compression algorithm, as implemented in programs that “zip” our computer files. I have and use WinZip v. 8.1 on my PC, but the particular implementation is not important; what’s important is the algorithm. Suppose the algorithm is fixed once and for all, for this definition of randomness, and we disallow any tinkering with it — otherwise we must understand that we tinker with our definition. It suffices that the algorithm does a pretty good job in attempting to compress any file (which is merely a sequence of bytes, i.e., numbers).
Having fixed the algorithm, my definition of randomness is a function that takes as input any sequence of numbers (which can also be the coded form of a configuration of dots, or molecules, in space), and outputs a number in the interval [0, 1), which is proportional to the percent of compression achieved. (Divide the percent by 100 to convert it to the interval [0, 1).) The parenthesis after 1 means that the number can never be exactly 1, because that would mean the sequence would vanish completely, and such total compression is inconceivable. But the closer the number is to 0, the more “random” the input sequence is. The bracket before 0 means that a compression of exactly 0 is possible, and it means “No compression at all was achieved”.
All right, I admit the above definition of randomness is not useful at all for theoretical math purposes, since it relies on a commercial algorithm — or on some compression algorithm in general, a specific one — and so it is algorithm-dependent. But my purpose was not to provide a theoretically useful definition, but to show that a definition can exist; it’s simply not true that randomness is an indefinable notion.
Thus we see that, in this definition, “random” is not a black-or-white notion (it’s not that a sequence either is random or is not), but has a gradation: a sequence (or configuration) can be “more random” than another one, which can in turn be “more random” than a third sequence, and so on. Nor does the definition require human cleverness, since it is all done by a fixed algorithm. Of course, an intelligent human might come and say, “Now look, this sequence that you declared ‘quite random’ is really quite compressible, thus not as random as you think, because I can use the such-and-such rule by which it is compressed quite a lot.” Fine. That means merely that the said person did not abide by our definition, but used a different approach, a different algorithm for achieving compression. One can always imagine a different algorithm. For instance, consider again the disordered dots on the right-side of the most recent figure (or the one below). One can state that the coordinates on which those dots stand are precisely the coordinates that, in that person’s numbering of coordinates, correspond to an N x N matrix, and thus the dots are perfectly ordered. The person altered the algorithm by which we number coordinates, and thus arrived at a non-random configuration. But that’s not impressive at all. The point is, given a fixed compression algorithm, how much can a sequence be compressed? The less it is, the more random it is declared to be, with respect to the given compression algorithm.
To be meaningful, the above definition depends on the assumption that the algorithm indeed performs reasonably well in compressing its input. But most commercial algorithms do have this feature. For example, consider the following three snapshots from an earlier version of our molecule-dispersal simulation, which used the same color to paint all molecules against a white background: